
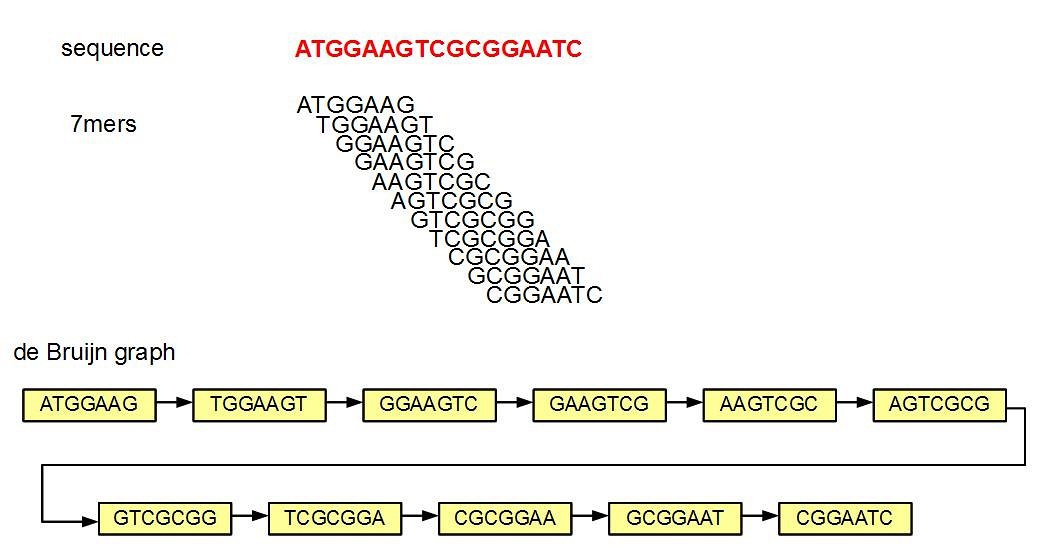
Пример выделения всех k-меров длины для k=7 из произвольной последовательности
© Изображение представлено пресс-службой МФТИ
МОСКВА, 2 марта. /ТАСС/. Ученые из НИИ Физико-химической медицины ФМБА РФ, МФТИ и университета ИТМО предложили новый метод сравнения метагеномов — совокупности последовательностей ДНК всех организмов в исследуемом биологическом материале. Он позволяет сравнивать образцы быстрее и эффективнее, чем существующие аналоги, сообщает пресс-служба МФТИ.
«Разработанная методика позволит более точно находить отличия между метагеномами разнообразных бактериальных сообществ, что, в частности, может помочь в изучении, диагностике и лечении многих заболеваний человека», — говорится в пресс-релизе.
Бактерии в человеческом организме
Бактериальных клеток в организме человека в десятки раз больше, чем собственных, причем большая часть из них находится в кишечнике. При этом состав бактериального сообщества влияет на риск возникновения заболеваний, выбор оптимального режима питания, настроение и даже творческие способности, а сами микроорганизмы чутко реагируют на все процессы в человеческом теле. Поэтому по кишечным метагеномам людей можно в перспективе оценивать риск диабета, различных воспалений кишечника и других заболеваний.
Традиционный подход в таком анализе — это сравнение по таксономическому составу (процентным долям каждого найденного вида микробов). Чтобы определить состав образца, его расшифрованные геномные последовательности сопоставляют с базой известных бактериальных геномов, называемых референсным набором. Однако такой подход имеет ряд недостатков. Во-первых, референсные геномы зачастую неточны: для изолированных в лаборатории видов они могут, например, нести набор генов, существенно отличающийся от того же вида в естественной среде. Во-вторых, не для всех организмов в принципе существуют собранные референсные геномы и поэтому часть последовательностей, для которых не находят соответствий в базе, часто просто не учитывается в анализе.
Сопоставление k-меров
В своей работе российские ученые использовали другой метод, основанный уже на сопоставлении k-меров: он не требует референсов и какой-нибудь дополнительной информации об исследуемых организмах, а также не игнорирует в анализе никакие последовательности. В основе метода лежит представление о геноме как о наборе всех встречающихся в нем нуклеотидных «слов», заданной длины k, называемых k-мерами. Поскольку геном является уникальной для каждого организма последовательностью, то и наборы таких «слов» различаются между отдельными организмами, а набор всех k-меров метагенома можно рассматривать как совокупность наборов, входящих в его состав организмов. Это позволяет судить о различиях в бактериальном составе при сравнении образцов между собой.
Для проверки эффективности k-мерной методики были использованы два набора метагеномных данных. Одни были реальными (кишечные метагеномы жителей США и Китая), а другие сгенерировали искусственно для проверки метода. В результате подход k-меров показал лучшие результаты, чем традиционное сопоставление с референсным набором: в метагеномах американцев и китайцев были найдены характерные и ожидаемые отличия, а искусственный метагеном был расшифрован с приемлемой точностью.
Генетическое исследование состава микрофлоры и подбор индивидуального метода лечения или профилактики болезней — это один из главных подходов персонализированной медицины будущего. Уже сейчас в исследованиях показано, что состав микробиоты может влиять на развитие ожирения или, например, аутизма.
Работа ученых из НИИ Физико-химической медицины ФМБА РФ, МФТИ и университета ИТМО опубликована в журнале BMC Bioinformatics.
Кстати, а вы знали, что на «Сделано у нас» статьи публикуют посетители, такие же как и вы? И никакой премодерации, согласований и разрешений! Любой может добавить новость. А лучшие попадут в наш Телеграм @sdelanounas_ru. Подробнее о том как работает наш сайт здесь👈
Другие публикации по теме
- Научный руководитель Института биохимии и генетики УФИЦ РАН Эльза Хусн...ентированной геномной селекции новых сортов растений и пород животных.
- Центр генетических исследований открыли в Москве. Ученые приступили к работе в виварии МГУ.
- Компания ГЕНЕРИУМ получила регистрационное удостоверение на медицинско...ами у людей с иммунодефицитными заболеваниями и состояниями.



Поделись позитивом в своих соцсетях
Комментарии 0